
Paid To Click


Custom Search
Tuesday, March 29, 2011
Microarray Method for Genetic Testing
DNA Sequencing Method: Cycle Sequencing
To sequence a piece of dna you need 1)a Template DNA 2) a short DNA primer that is complementary to the dna you want to sequence, 3)A enzyme called DNA polymerase,(4) Four nucleotides.(A,C,G,T), To this mix ,we also add a second type of nucleotide; one that has a slightly different chemical formula, These dideoxynucleotides(diddtp) can be recognized by a DNA sequencer.
To start the sequencing reaction this mixture is heated to 96C ,so the template DNA's two complementary strand separates,Then the temperature is lowered, so that the short "primer" sequence finds its complementary sequence in the template DNA.Finally the temperature is raised 60c,this allows the enzyme to bind to the DNA and create a new strand of DNA.
The sequence of this new DNA is complementary to the original DNA strand. The enzyme makes no distinction between dNTPs or didNTPs.each time a didNTP is incorporated, in this case didATP,The synthesis stops. Because billion of DNA molecules are present in the test tube, the strand can be terminated at any position. This results in collection of DNA strands of many different lengths.
The sequencing reaction is transferred from the test tube to a lane of a polyacrylamide gel. The gel is placed into a DNA sequencer for electrophoresis and analysis. The fragments migrate according to size and each is detected as it passes a laser beam at the bottom of the gel. Each type of dideoxynucleotide emits colored light of a characteristic wavelength and is recorded as a colored band on a simulated gel image, and finally computer program interprets the raw data and outputs an electropherogram with colored peaks representing each letter in the sequence.the sequence fragments are sorted out according to the size, starting from the shortest to longest one, the stimulated gel image is read from bottom to top, starting with the smallest fragment, Thus we sequences present in template DNA.
Wednesday, March 23, 2011
UniSeq DNA Sequencing System
UniSeq™ is a universal DNA sequencing primer walking technology developed by Nucleics for large scale DNA sequencing applications like whole genome sequencing projects. The UniSeq DNA sequencing library & software offers the following advantages over other sanger DNA sequencing methodologies:
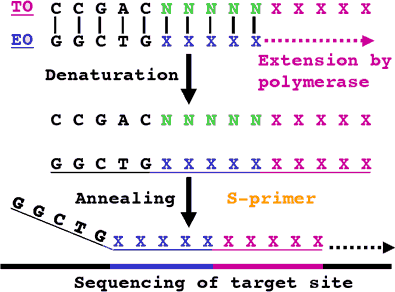
Figure 1. Generation of template specific UniSeq S-primer. The EO hybridizes to the TO and gets extended to form the S-primer. N: degenerated oligonucleotide positions. X: specific positions of variable nucleotide value.
By selecting specific EO and TO oligonucleotides from a small, pre-synthesised library of 768 oligonucleotides, over 131,000 different, template specific primers can be created. This simple combinatorial effect forms the basis for the high specificity and universal applicability of the UniSeq system in DNA sequencing.
The UniSeq system is fully compatible with the common DNA sequencing reagents (e.g. BigDye™ from Applied Biosystems or DYEnamic™ from Amersham Biosciences) and gives excellent results using modern capillary DNA sequencers (Figure 2).
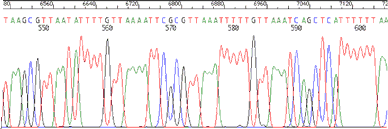
Figure 2. UniSeq reaction sequencing trace. Two hundred nanograms of plasmid DNA was sequenced with BigDye vers.3.1 and UniSeq primers EO 18 and TO 447. The DNA sequencing reaction was purified by ethanol precipitation and analyzed on an ABI 3700 DNA sequencer.
The UniSelect™ DNA sequencing software is provided for the automated and optimized selection of the optimal EO & TO primer pairs for each DNA template. The UniSelect software also supplies an automated interface to control the robotic pipetting of the UniSeq oligonucleotides, DNA templates and other required sequencing reagents.
To aid the finishing of Whole Genome Sequencing (WGS) projects, an additional software package (UniFinish™) is available. UniFinish is able to parse ACE format assembly files and select the EO/TO oligonucleotides, together with the appropriate DNA templates, required to close non-physical DNA sequence gaps in the genome assembly.
UniSeq provides the advantages of both WGS strategies. It offers the speed and simplicity of the RSS approach, while providing the data efficiency inherent with the PWS approach. Computer simulations and limited trials have shown that UniSeq DNA sequencing system offers cost and time savings of greater than 80% over current WGS approaches.
- A fast and cost effective universal primer walking approach
- Very simple and robust protocol
- Compatible with all Sanger DNA sequencing technologies and equipment
- Fully adaptable to high throughput, large scale DNA sequencing facilities
The UniSeq DNA sequencing system
The UniSeq DNA sequencing system utilizes a unique methodology to reliably create template specific DNA sequencing primers. The process of generating each UniSeq DNA sequencing primer involves the addition of an "E"- and "T"-oligonucleotide, together with a specifically formulated additive mixture, directly to the sequencing reaction. These oligonucleotides (termed EO and TO) hybridize during the sequencing reaction to produce the template specific primer (S-primer) (Figure 1).Figure 1. Generation of template specific UniSeq S-primer. The EO hybridizes to the TO and gets extended to form the S-primer. N: degenerated oligonucleotide positions. X: specific positions of variable nucleotide value.
By selecting specific EO and TO oligonucleotides from a small, pre-synthesised library of 768 oligonucleotides, over 131,000 different, template specific primers can be created. This simple combinatorial effect forms the basis for the high specificity and universal applicability of the UniSeq system in DNA sequencing.
The UniSeq system is fully compatible with the common DNA sequencing reagents (e.g. BigDye™ from Applied Biosystems or DYEnamic™ from Amersham Biosciences) and gives excellent results using modern capillary DNA sequencers (Figure 2).
Figure 2. UniSeq reaction sequencing trace. Two hundred nanograms of plasmid DNA was sequenced with BigDye vers.3.1 and UniSeq primers EO 18 and TO 447. The DNA sequencing reaction was purified by ethanol precipitation and analyzed on an ABI 3700 DNA sequencer.
The UniSelect™ DNA sequencing software is provided for the automated and optimized selection of the optimal EO & TO primer pairs for each DNA template. The UniSelect software also supplies an automated interface to control the robotic pipetting of the UniSeq oligonucleotides, DNA templates and other required sequencing reagents.
To aid the finishing of Whole Genome Sequencing (WGS) projects, an additional software package (UniFinish™) is available. UniFinish is able to parse ACE format assembly files and select the EO/TO oligonucleotides, together with the appropriate DNA templates, required to close non-physical DNA sequence gaps in the genome assembly.
Feature and benefits of UniSeq
Genomes are finished faster
The UniSeq DNA sequencing system provides a competitive alternative to DNA sequencing with custom synthesized oligonucleotides. While many advances have been made in the automation of oligonucleotide synthesis, this process is still complex (eg. requiring high maintenance machinery) and slow (several hours per synthesis). These limitations have become extremely critical for high throughput DNA sequencing facilities where modern capillary DNA sequencers have reduced the separation time to one to two hours. The generation of specific DNA sequencing primers by the UniSeq process during the DNA sequencing reaction step (i.e. no time-costs) breaks the current bottleneck imposed by custom oligonucleotide synthesis and allows for the most efficient utilization of existing DNA sequencing instruments and other equipment.Lower per finished base costs
Currently, two major strategies are used in WGS – Random Shotgun DNA Sequencing (RSS) and Primer Walking DNA Sequencing (PWS). Most WGS projects currently employ RSS, especially in early stages of projects. However, RSS requires generating a large amount of redundant DNA sequence (commonly 6 to 15 times the genome size) for assembly purposes. The alternative PWS strategy requires relatively little redundant DNA sequence data, however, it requires a large numbers of custom made oligonucleotide primers. The high costs (US$3 to 5 per primer), together with the required synthesis delay, have prevented the general adoption of PWS strategy in WGS Genomics projects.UniSeq provides the advantages of both WGS strategies. It offers the speed and simplicity of the RSS approach, while providing the data efficiency inherent with the PWS approach. Computer simulations and limited trials have shown that UniSeq DNA sequencing system offers cost and time savings of greater than 80% over current WGS approaches.
Increased DNA sequencing flexibility
Nucleics has extensively tested UniSeq in house in a number of general DNA sequencing and WGS projects. In addition, Nucleics has formulated novel strategies for the easy and smooth implementation of UniSeq into industrial scale DNA sequencing facilities. We also offer consulting services to help integrate UniSeq into new or existing DNA sequencing facilities.DNA Sequencing: Shotgun Sequencing
Shotgun Sequencing was first developed by Craig Ventor in 1996. He developed it as he was working in the Genome Research Institute. It was him who also made it popular. He then went on to start his Celera Corporation with the sole mission and goal of doing the sequencing of mainly the human genome in as little as three years. But of course, some say that in genetics, the shotgun sequencing was in fact first developed by the double Nobel laureate, Fred Sanger, in the 1970s .
Shotgun Sequencing is a DNA sequencing method that involves the physically breaking down of a long stretch of DNA into very small fragments; about 2,000 base-pair. These fragments are then cloned, sequenced and also assembled with the use of computer analysis.
Also known in other circles and by other people as shotgun cloning, this method is said to be one of the harbinger technologies which is mainly responsible for bringing about what we now have as full genome sequencing. The sequencing of the human genome was done by Craig Ventor’s Celera Corporation as well as by the Human Genome Project. They used a map-based sequencing while Craig’s Ventor’s Celera used shotgun sequencing.
Today, however, Craig Ventor’s Shotgun Sequencing is the preferred system and method for doing other types of genome sequencing. And as science and technology improves, it’s clearly obvious that more things will be discovered that will alter or improve the method, for the better. For example, there is now the method called the next-generation sequencing, which is said to result in high coverage than Craig’s. As they say, only time will tell what the future has for this and other methods yet unravelled because man is discovering newer things each and every day. And as many continue to share what they know with others, better methods will obviously be discovered.
Lehninger's Principles of Biochemistry - 5th Edition Ebook Download

Sunday, March 20, 2011
Separation of DNA Fragments Using PAGE Method
This method is able to separate DNA fragments with the size of as small as 10 bp and up to 1 kb with the resolution of as little as 1 bp. While agarose gel electrophoresis is only able to separate DNA fragments with the bigger size that PAGE does or in the size range of 100 nucleotides to around 10 – 15 kb.
Materials:
Materials:
- Gel apparatus: Many designs of apparatus are commercially available. The gel is poured between two vertical plates held apart by spacers
The plates should be cleaned thoroughly and then wiped with ethanol. To help ensure that the gel only sticks to one plate when the apparatus is disassembled, apply silicon to one of the gel plates. This is easily done by wiping the plate with a tissue soaked in dimethyl dichlorosilane solution and then washing the plate in distilled water followed by ethanol. If the plates are baked at 100oC for 30 min, the siliconization will last four to five gel runs.
- Deionized H2O: Autoclaved water is not necessary for the gel mix or running buffer, but it should be used for diluting samples and purification from gel slices.
- l0x TBE: 108 g of Trizma base (Tris), 55 g of boric acid, and 9.3 g of ethylenediaminetetraacetic acid (EDTA) (disodium salt). Make up to I L solution with deionized H2O, which should be discarded when a precipitate forms.
- Acrylamide stock: 30% acrylamide, 1% N,N'-methylene bisacrylamide. Store at 4oC. This is available commercially, or it can be made by dissolving acrylamide and bisacrylamide in water, which should be filtered. Acrylamide is a neurotoxin and therefore must be handled carefully. Gloves and a mask must be worn when weighing out.
- APS: 10% Ammonium persulphate (w/v). This can be stored at 4oC for 1-2 months.
- TEMED: N,N,N',N'-tetramethyl-l,2-diaminoethane. Store at 4oC.
- 5X sample buffer: 15% Ficoll solution, 2.5X TBE, 0.25% (w/v) xylene cyanol and 0.025% (w/v) bromophenol blue.
- Ethidium bromide: A l0-mg/mL solution. Ethidium bromide is a potent mutagen and should be handled with care. Store at 4oC in the dark.
- For 50 mL, enough for a 18 x 14 x 0.15 cm gel, mix l0x TBE, acrylamide, H2O, and APS as described in Table below.
- Just prior to pouring, add 50 microliters of TEMED and mix by swirling.
- Immediately pour the gel mix between the gel plates and insert the gel comb. Leave to set; this takes about 30 min.
- Fill the gel apparatus with 0.5X TBE and remove the comb. Use a syringe to wash out the wells, this may take multiple washes. It is important to remove as much unpolymerized acrylamide as possible because this impairs the running in of the samples
If you are separating very small fragments, e.g., less than 50 bp, the gel should be prerun for 30 min, as this elevates the resolution problem experienced with fragments running close to the electrophoresis front..
- Add 0.2 volume of 5x sample buffer to each sample, usually in 10-20 microliters of TE, water, or enzyme buffer. Mix and spin the contents to the bottom of the tube
High-salt buffers (above 50 mM NaCl) will affect sample mobility and tend to make bands collapse. In this case, salt should be removed by ethanol precipitation..
- Load the samples on the gel and run at 200-300 V (approximately 10 V/cm) until the bromophenol blue band is two-thirds of the way down the gel; this takes about 2.5 h
Do not run the gel faster than 10 V/cm, as this will cause the gel to overheat, affecting the resolution. The gel can be run more slowly, e.g., 75 V will run overnight.
- Disassemble the gel apparatus and place the gel to stain in I mg/mL of ethidium bromide for approximately 30 min. View the stained gel on a transilluminator.

Monday, March 14, 2011
Purification Of Plasmid DNA
After the initial characterization, it is possible to purify further some or all of the plasmid DNAs by RNase digestion and extraction with organic solvents. This further purified DNA is suitable for techniques such as DNA sequencing, subcloning or the production of gene probes. In order to purify plasmid DNA after the isolation process, any residual RNA and contaminating protein are removed. This purification step involves two main steps, which are, first, removing residual RNA by using RNase in order to digest RNA and, second, extract contaminating protein using organic solvents, phenol-chloroform.
Materials:
Materials:
- RNase A: Make up as a solution in water at 10 mg/mL, Heat for 10 min in a boiling water bath or heating block to eliminate any DNase activity. Aliquot and store at -20oC.
- 0.4 M Ammonium acetate.
- Chloroform= A 24: 1 mix of chloroform and isoamyl alcohol. Store at 4oC.
- Phenol/chloroform= 25:24: 1 mix of TE-equilibrated phenol, chloroform, and isoamyl alcohol. Store at 4oC.
- 100% Ethanol.
- Sterile wooden toothpicks.
- Add 50 microliters of 4 M ammonium acetate containing 200 micrograms/mL RNase A to each miniprep and incubate it at room temperature for 20 min.
- Add 100 microliters of phenol/chloroform to each DNA preparation.
- Vortex briefly and centrifuge at high speed for 2 min in a microfuge. Remove the top layer containing the DNA and place it in a new sterile tube.
- Add 100 microliters of chloroform to each tube.
- Vortex briefly and centrifuge at high speed in a microfuge for 2 min. Remove the DNA in the top layer and place it in a second sterile tube.
For phenol/chloroform extractions avoid removing material from the interface.
- Add 200 microliters of 100% ethanol to each tube.
- Shake briefly to precipitate the DNA and centrifuge at high speed for 5 min at room temperature.
Friday, March 11, 2011
The Vertical Farm
The Problem



Advantages of Vertical Farming
By the year 2050, nearly 80% of the earth's population will reside in urban centers. Applying the most conservative estimates to current demographic trends, the human population will increase by about 3 billion people during the interim. An estimated 109 hectares of new land (about 20% more land than is represented by the country of Brazil) will be needed to grow enough food to feed them, if traditional farming practices continue as they are practiced today. At present, throughout the world, over 80% of the land that is suitable for raising crops is in use (sources: FAO and NASA). Historically, some 15% of that has been laid waste by poor management practices. What can be done to avoid this impending disaster?
A Potential Solution: Farm Vertically
The concept of indoor farming is not new, since hothouse production of tomatoes, a wide variety of herbs, and other produce has been in vogue for some time. What is new is the urgent need to scale up this technology to accommodate another 3 billion people. An entirely new approach to indoor farming must be invented, employing cutting edge technologies. The Vertical Farm must be efficient (cheap to construct and safe to operate). Vertical farms, many stories high, will be situated in the heart of the world's urban centers. If successfully implemented, they offer the promise of urban renewal, sustainable production of a safe and varied food supply (year-round crop production), and the eventual repair of ecosystems that have been sacrificed for horizontal farming.
It took humans 10,000 years to learn how to grow most of the crops we now take for granted. Along the way, we despoiled most of the land we worked, often turning verdant, natural ecozones into semi-arid deserts. Within that same time frame, we evolved into an urban species, in which 60% of the human population now lives vertically in cities. This means that, for the majority, we humans are protected against the elements, yet we subject our food-bearing plants to the rigors of the great outdoors and can do no more than hope for a good weather year. However, more often than not now, due to a rapidly changing climate regime, that is not what follows. Massive floods, protracted droughts, class 4-5 hurricanes, and severe monsoons take their toll each year, destroying millions of tons of valuable crops. Don't our harvestable plants deserve the same level of comfort and protection that we now enjoy? The time is at hand for us to learn how to safely grow our food inside environmentally controlled multistory buildings within urban centers. If we do not, then in just another 50 years, the next 3 billion people will surely go hungry, and the world will become a much more unpleasant place in which to live. Advantages of Vertical Farming
- Year-round crop production; 1 indoor acre is equivalent to 4-6 outdoor acres or more, depending upon the crop (e.g., strawberries: 1 indoor acre = 30 outdoor acres)
- No weather-related crop failures due to droughts, floods, pests
- All VF food is grown organically: no herbicides, pesticides, or fertilizers
- VF virtually eliminates agricultural runoff by recycling black water
- VF returns farmland to nature, restoring ecosystem functions and services
- VF greatly reduces the incidence of many infectious diseases that are acquired at the agricultural interface
- VF converts black and gray water into potable water by collecting the water of
evapotranspiration - VF adds energy back to the grid via methane generation from composting non-edible
parts of plants and animals - VF dramatically reduces fossil fuel use (no tractors, plows, shipping.)
- VF converts abandoned urban properties into food production centers
- VF creates sustainable environments for urban centers
- VF creates new employment opportunities
- We cannot go to the moon, Mars, or beyond without first learning to farm indoors on
earth - VF may prove to be useful for integrating into refugee camps
- VF offers the promise of measurable economic improvement for tropical and subtropical
LDCs. If this should prove to be the case, then VF may be a catalyst in helping to reduce or even reverse the population growth of LDCs as they adopt urban agriculture as a strategy for sustainable food production. - VF could reduce the incidence of armed conflict over natural resources, such as water
and land for agriculture
Urban Farming Grows Up
Urban Farming’s mission is to create an abundance of food for people in need by planting, supporting and encouraging the establishment of gardens on unused land and space while increasing diversity, raising awareness for health and wellness, inspiring and educating youth, adults and seniors to create an economically sustainable system to uplift communities around the globe.
Friday, March 4, 2011
INDIRECT ELISA
INDIRECT ELISA :
INDIRECT ELISA The indirect ELISA utilizes an unlabeled primary antibody in conjunction with a labeled secondary antibody. Since the labeled secondary antibody is directed against all antibodies of a given species (e.g. anti-mouse), it can be used with a wide variety of primary antibodies (e.g. all mouse monoclonal antibodies).INDIRECT ELISA :
INDIRECT ELISA Advantages of indirect detection Wide variety of labeled secondary antibodies are available commercially. Versatile, since many primary antibodies can be made in one species and the same labeled secondary antibody can be used for detection. Immunoreactivity of the primary antibody is not affected by labeling. Sensitivity is increased because each primary antibody contains several epitopes that can be bound by the labeled secondary antibody, allowing for signal amplification.
DIRECT ELISA

DIRECT ELISA :
DIRECT ELISA The direct ELISA uses the method of directly labeling the antibody itself. Microwell plates are coated with a sample containing the target antigen, and the binding of labeled antibody is quantitated by a colorimetric, chemiluminescent, or fluorescent end-point.DIRECT ELISA :
DIRECT ELISA Advantages of Direct Detection Quick methodology since only one antibody is used. Cross-reactivity of secondary antibody is eliminated. Disadvantages of Direct Detection Immunoreactivity of the primary antibody may be reduced as a result of labeling. Labeling of every primary antibody is time-consuming and expensive. No flexibility in choice of primary antibody label from one experiment to another. Little signal amplification.COMPETITIVE ELISA
COMPETITIVE ELISA :
COMPETITIVE ELISA In this Unlabeled antibody is incubated in the presence of its antigen. These bound antibody/antigen complexes are then added to an antigen coated well. The plate is washed unbound antibody is removed. The secondary antibody, specific to the primary antibody is added. This second antibody is coupled to the enzyme. A substrate is added, and remaining enzymes elicit a chromogenic or fluorescent signal. For competitive ELISA, the higher the original antigen concentration, the weaker the eventual signal.
ELISA : Enzyme-Linked ImmunoSorbent Assay
How the Test is Performed
Blood is typically drawn from a vein, usually from the inside of the elbow or the back of the hand. The site is cleaned with germ-killing medicine (antiseptic). The health care provider wraps an elastic band around the upper arm to apply pressure to the area and make the vein swell with blood.Next, the health care provider gently inserts a needle into the vein. The blood collects into an airtight vial or tube attached to the needle. The elastic band is removed from your arm.
Once the blood has been collected, the needle is removed, and the puncture site is covered to stop any bleeding.
In infants or young children, a sharp tool called a lancet may be used to puncture the skin and make it bleed. The blood collects into a small glass tube called a pipette, or onto a slide or test strip. A bandage may be placed over the area if there is any bleeding.
The sample is sent to a laboratory where the targeted antibody (or antigen) is linked to an enzyme. If the target substance is in the sample, the test solution turns a different color.
How to Prepare for the Test
No special preparation is needed.How the Test Will Feel
When the needle is inserted to draw blood, some people feel moderate pain, while others feel only a prick or stinging sensation. Afterward, there may be some throbbing.Why the Test is Performed
This test is often used to see if you have been exposed to viruses or other substances that cause infection. It is often used to screen for current or past infections.Normal Results
Normal values depend on the type of substance being identified. Normal value ranges may vary slightly among different laboratories. Talk to your doctor about the meaning of your specific test results.What Abnormal Results Mean
Abnormal values depend on the type of substance being identified. In some people, a positive result may be normal.Risks
Veins and arteries vary in size from one patient to another and from one side of the body to the other. Obtaining a blood sample from some people may be more difficult than from others.Other risks associated with having blood drawn are slight but may include:
- Excessive bleeding
- Fainting or feeling light-headed
- Hematoma (blood accumulating under the skin)
- Infection (a slight risk any time the skin is broken)
Alternative Names
Enzyme-linked immunoassay; EIAReferences
Ashihara Y, Kasahara Y, Nakamura RM. Immunoassay and immunochemistry. In: McPherson RA, Pincus MR, eds. Henry's Clinical Diagnosis and Management by Laboratory Methods. 21st ed. Philadelphia, Pa: Saunders Elsevier; 2006:chap 43.
Subscribe to:
Posts (Atom)